
From No to Know: Translating Regulatory Setbacks into Competitive Advantages
Authored by Cassandra Carroll, PhD; Kaitlyn Delano, PhD; Kinza Maxood, MD; Vrudhi Shah, MSc; Margot Arntfield, PhD; JJ Mifsud
Listen to this article
What you’ll learn:
Our analysis of 202 CRL files illuminates the hidden drivers of FDA rejections, offering companies a rare opportunity to learn from past missteps and strengthen future submissions.
Manufacturing issues surfaced as the leading cause of CRLs across therapeutic areas, while clinical and labeling concerns also emerged as significant and often overlapping hurdles.
The findings highlight the value of early, cross-functional alignment and the strategic advantage of combining regulatory expertise with tools that can turn agency feedback into foresight.
Turning 202 Setbacks into Strategy: What the FDA’s Letters Reveal
Before a product can reach the market, it must clear the FDA’s final regulatory hurdle. When that doesn’t happen, the agency issues a Complete Response Letter (CRL)—a formal notification that the application, as submitted, cannot be approved. And that “no” carries real downstream consequences: delayed approvals can translate into significant financial setbacks for companies and, more critically, prolonged access gaps for patients still waiting on new treatment options. Beyond those setbacks and the surface-level “no,” CRLs often contain a wealth of insight—if you know where to look.
Historically, CRLs have functioned as something of a Black Box. Like Untitled and Warning letters, CRLs provide a window into the agency’s evolving priorities. Shared privately with sponsors and only occasionally released in redacted form, CRLs offer limited transparency to the broader industry. Yet, much like Black Box warnings themselves, they often highlight serious, foundational concerns such as chemistry, manufacturing, and control (CMC) issues, unclear benefit-risk profiles, or unresolved safety issues.
The issues raised in CRLs aren’t typically insurmountable; however, they tend to require significant revision or program-level reevaluation to reach approval. Though CRLs are not official industry-guidance documents, they reflect the FDA’s real-time thinking; how it is interpreting established standards, where it is intensifying its scrutiny, and what it expects from sponsors in order to move forward.
On July 10, 2025, the FDA released 202 CRL files, representing a subset of all CRLs issued between 2020 and 2024 (including earlier CRLs dating back to 2007), specifically those associated with drugs that were ultimately approved.
The dataset, while reflecting only a select portion of CRLs issued during this time frame, offers the industry a rare opportunity to examine the rationale behind these regulatory setbacks and extract broader insights. These learnings can, and should, be treated as strategic roadmaps, helping teams anticipate and proactively address similar challenges in future submissions.

Learning from the No’s
When approached with the right expertise, these letters hold a treasure trove of valuable data. By combining regulatory-intelligence strategies with advanced proprietary AI tools, we worked quickly to uncover key trends, validate our findings, and extract strategic insights.
Here are just a fraction of what we can learn from these letters, with many more insights to follow.
Key Drivers of CRLs: Manufacturing, Clinical, and Labeling Issues
Before exploring specific deficiency categories, it’s important to understand the composition of the dataset.
Our analysis includes 284 individual CRLs issued across 202 application files, reflecting instances where a single application received multiple letters throughout its review cycle. Notably, 166 of these CRLs cited more than one type of deficiency, such as both clinical and CMC concerns, highlighting the layered nature of FDA feedback and explaining why category totals exceed 100%.
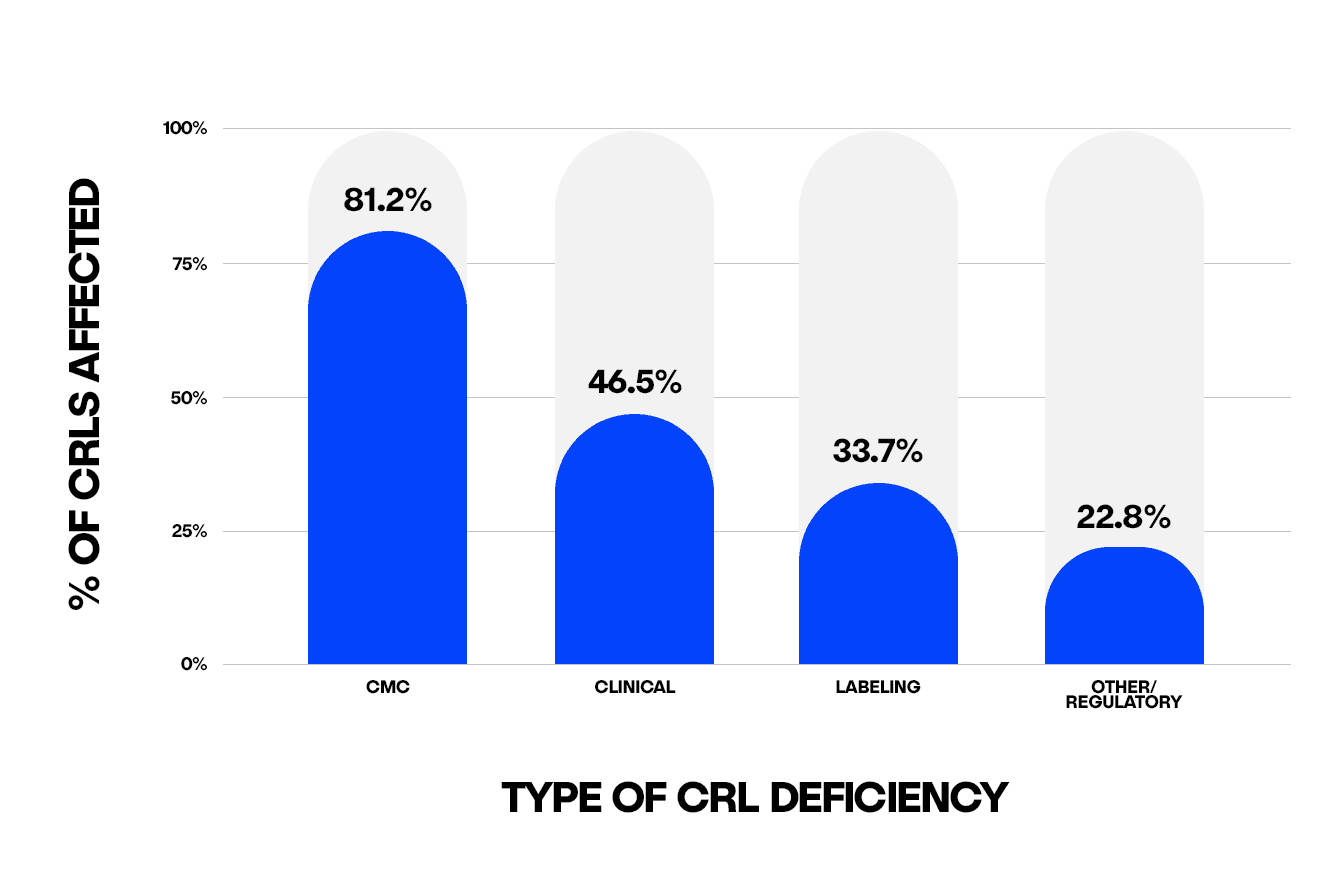
CMC Issues
CMC issues were the single largest driver of CRLs, with over 80% of unique CRLs cited within the manufacturing process. This broad category encompassed a range of issues including:
Facility inspections
Gaps in the manufacturing process
Concerns about product quality during production
In several cases, deficiencies included the following:
Absence of a robust, validated quality program to ensure critical product-quality attributes are maintained throughout manufacturing
Lack of adequate and appropriate controls to validate and monitor operating processes
Need to define the roles and responsibilities of the different facilities involved in the manufacturing of a combination product
While specific details were not always outlined for facility inspection-related deficiencies, the overarching CMC feedback often pointed to inadequate methods, facilities, or controls used for the manufacture, processing, packing, or holding of the drug substance or product—raising concerns about the drug’s identity, strength, quality, purity, stability, and bioavailability. It is worth noting, some CRLs issued between 2020 and 2022 explicitly cited the FDA’s inability to complete facility inspections due to COVID-19 travel restrictions, suggesting that CMC deficiencies during this period may be inflated due to pandemic-related limitations rather than systemic trends.
Within this dataset, the clear prominence of CMC-related issues is difficult to ignore and seems to suggest that technical execution and operational readiness may present greater barriers to approval than clinical performance. That said, because this analysis includes only drugs that were ultimately approved, it’s possible that applications receiving significant clinical feedback were not resubmitted, with sponsors opting to discontinue development rather than pursue the required updates.
Clinical Deficiencies
Clinical deficiencies were the second-most frequently cited category, appearing in 46.5% of CRLs, and could represent the most difficult and resource-intensive issues to resolve, sometimes going as far as requesting new clinical trials to resolve deficiencies.
Deficiencies spanned a range of concerns, including:
The lack of substantial evidence for effectiveness
Unsatisfactory risk-to-benefit profile
Unresolved safety signals that raised questions around approvability
In several cases, deficiencies stemmed from:
Study design limitations, including inappropriate dosage and/or administration
Confounding population factors
Incomplete subject follow-up or incomplete data collection, ultimately impeding the interpretation of the data and limiting the strength of conclusions that could be drawn
While not the most commonly cited issue in CRLs, the high impact and recurrence of clinical deficiencies underscore the importance of early and proactive alignment with the FDA during trial design. Formal regulatory review interactions with the FDA, such as pre-New Drug Application (NDA) or pre-Biologics License Application (BLA) discussions, provide critical opportunities to engage early and help reduce the risk of misalignment as well as build confidence in the program’s scientific and operational rigor.
Labeling Deficiencies
Labeling deficiencies, cited in approximately one-third of CRLs, typically involved changes to the Prescribing Information (PI), including the Medication Guide, the carton or container labels, or proprietary (brand) naming. While these issues may appear administrative and can be easily underestimated, they introduce downstream delays if not proactively addressed.
In many cases, PI and carton-label feedback was withheld until more critical deficiencies, such as clinical or CMC issues, could be addressed. Similarly, proprietary name evaluations were sometimes deferred or conditionally approved, pending the outcome of other outstanding issues in the application. These findings underscore the importance of treating labeling elements as integral, not secondary components of regulatory strategy.
Other/Regulatory
The Other/Regulatory category includes any concerns related to missing studies, particularly the quality of pharmacologic studies, regulatory procedures, or legal barriers such as exclusivity, and is cited in just under a quarter of CRLs. Having a clear and accurate understanding of what the FDA is expecting to see in these submissions may have avoided some of these challenges.
Ensuring a submission is right the first time requires early and intentional collaboration across regulatory, medical, and commercial teams, alongside proactive FDA engagement to align on expectations.
CRL Signals by Therapeutic Area: Knowing the Usual Suspects to Stay Ahead
When we look at the five therapeutic areas (TA) with the most CRLs in this dataset, manufacturing consistently remains the biggest roadblock, showing up in about 71% of letters in neurology and peaking in oncology at 97%. Neurology also carries a high clinical burden, with 64.3% of its letters citing issues related to trial design or endpoints. In comparison, labeling gaps appear in 50% of neurology CRLs and 43% of those in pain, highlighting added complexity in PI and packaging.
Early patterns suggest that each TA may face distinct regulatory pressure points. However, clinical and labeling challenges could be underrepresented, given that the dataset includes only products that were eventually approved. In addition, the higher CRL totals in oncology, cardiovascular, and dermatology may reflect both heavier filing volume and added clinical or regulatory complexity.
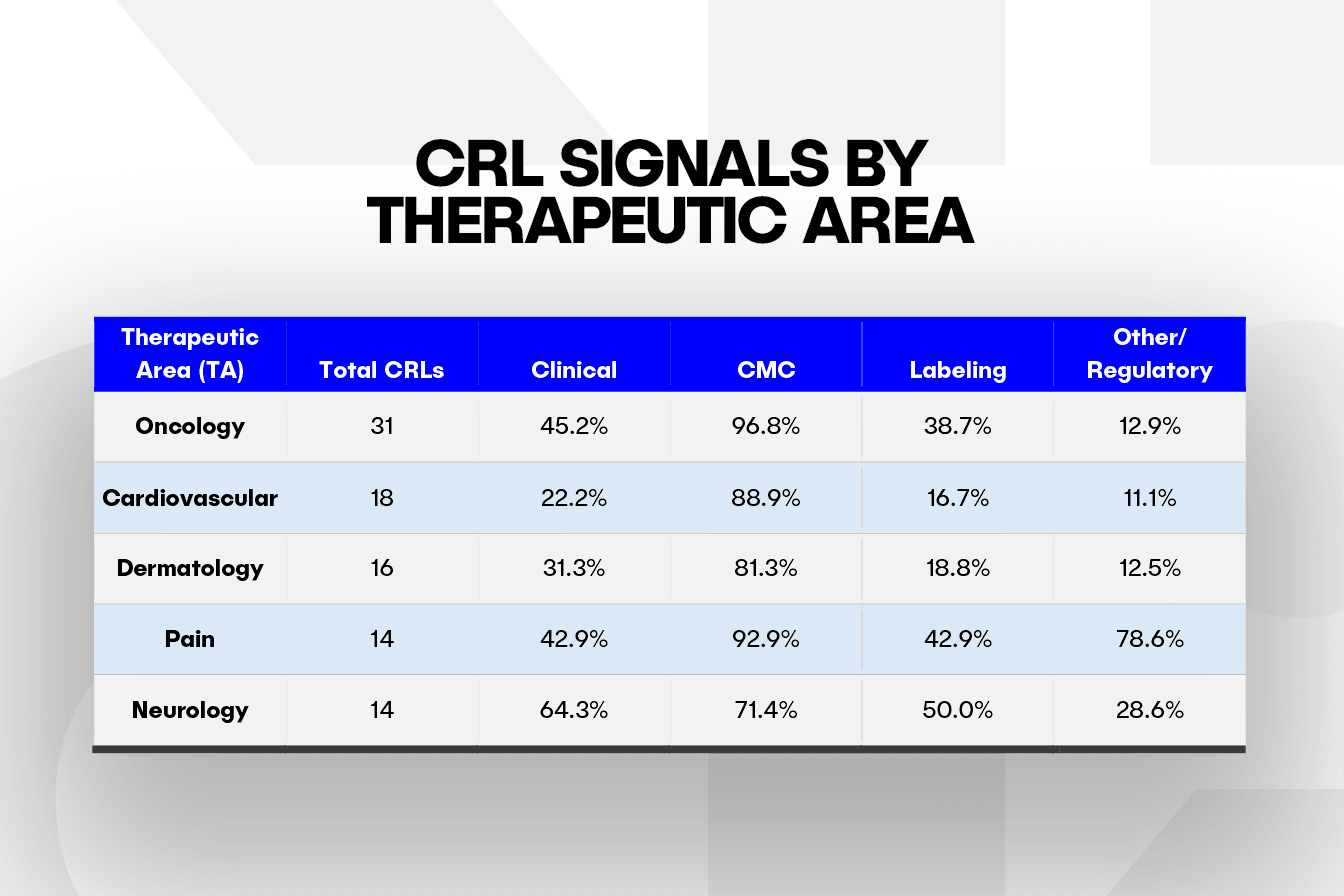
Spotting a TA’s typical bottleneck early, whether it involves labeling, risk evaluation and mitigation strategy (REMS) planning, or device-combo CMC, allows teams to bring the right specialists into the review cycle sooner and avoid preventable CRLs.
Uncovering Trends in BLA vs. NDA Submissions
When comparing concerns raised in CRLs from BLA submissions to NDA submissions, we observe some interesting trends that will warrant further analysis. In this set of CRLs, unique submissions for BLAs account for approximately a quarter of the total number, with NDA submissions representing the remaining three quarters. This distribution is consistent across clinical, CMC, and labeling concerns, suggesting BLA and NDA face similar challenges in these areas.
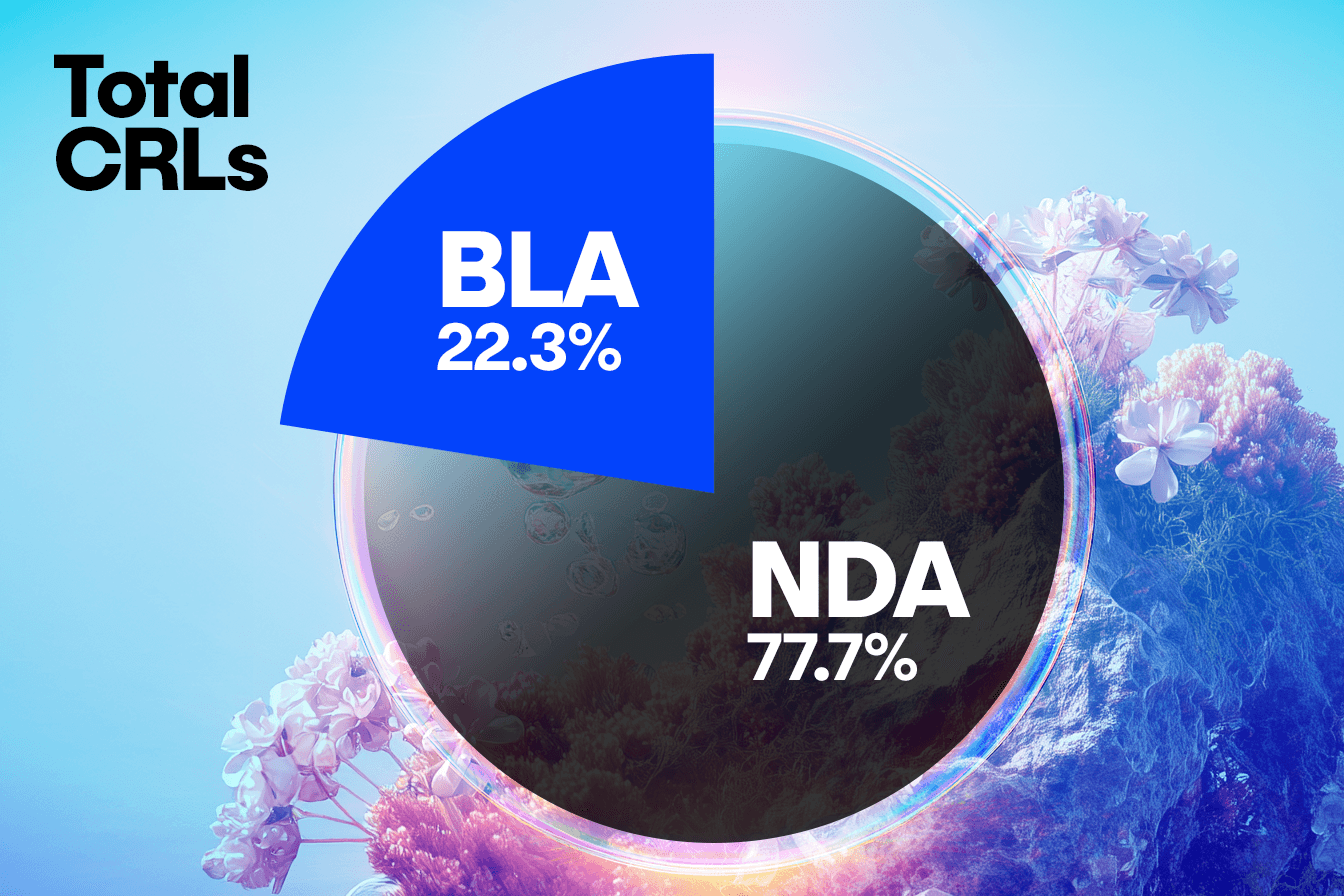
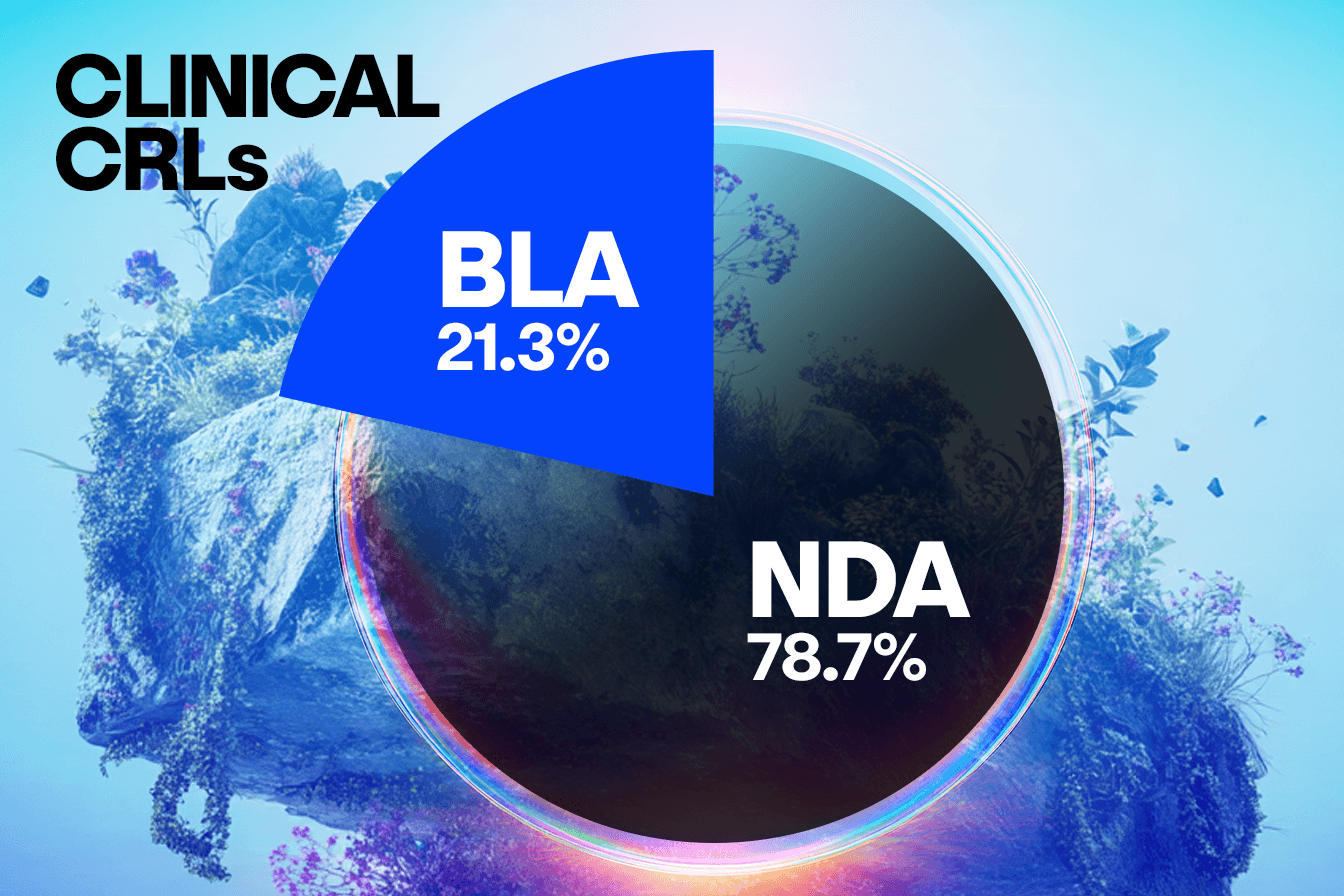
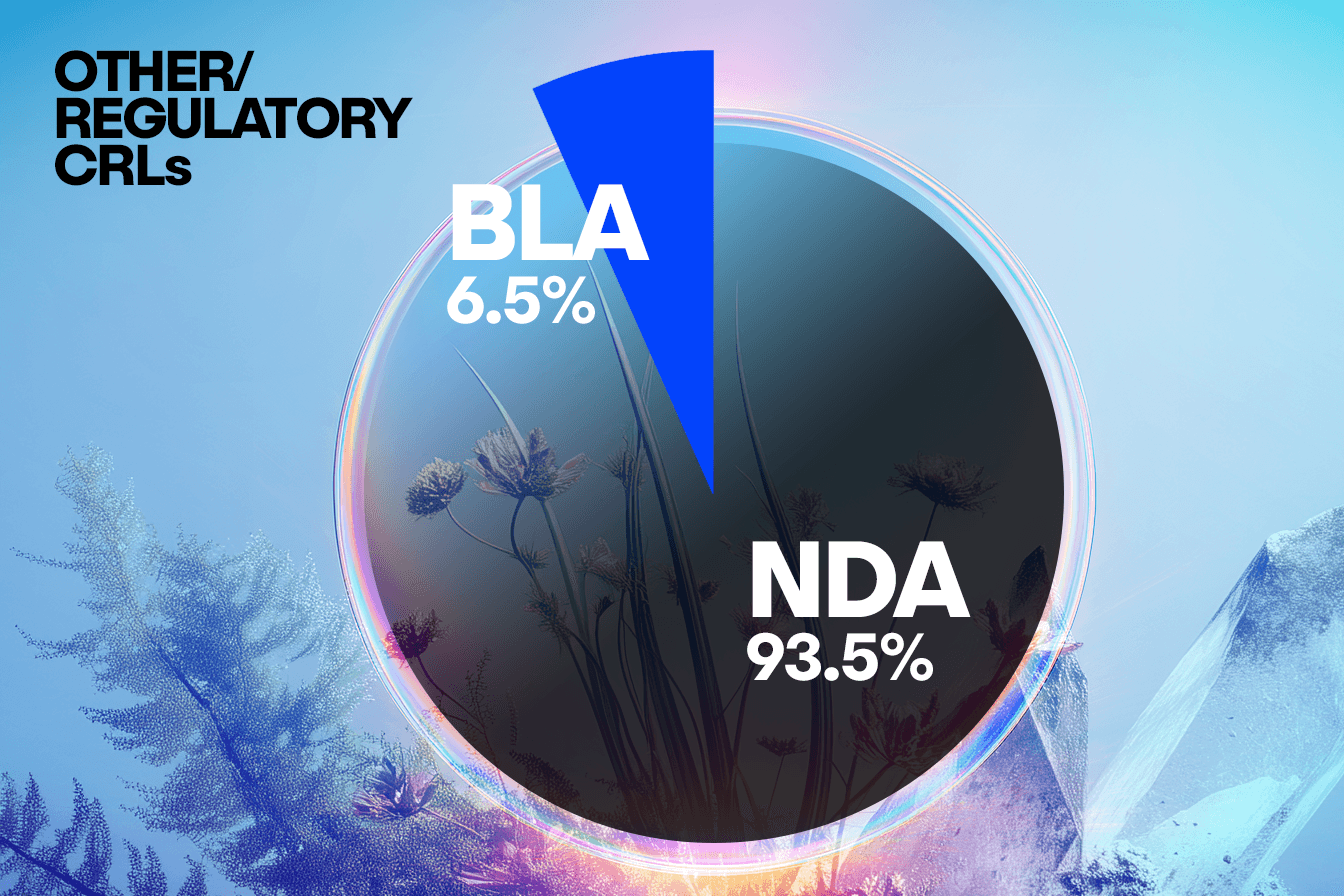
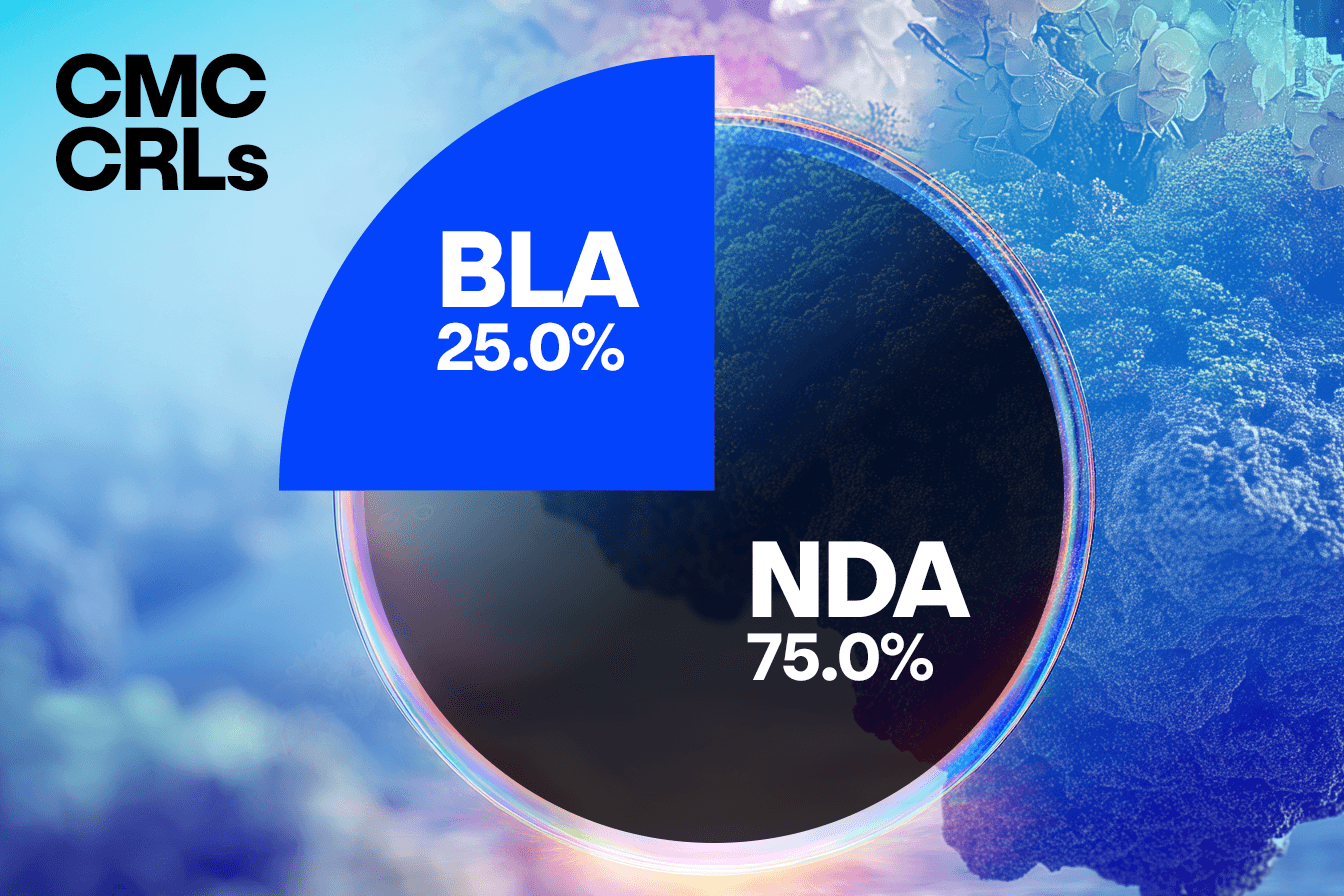
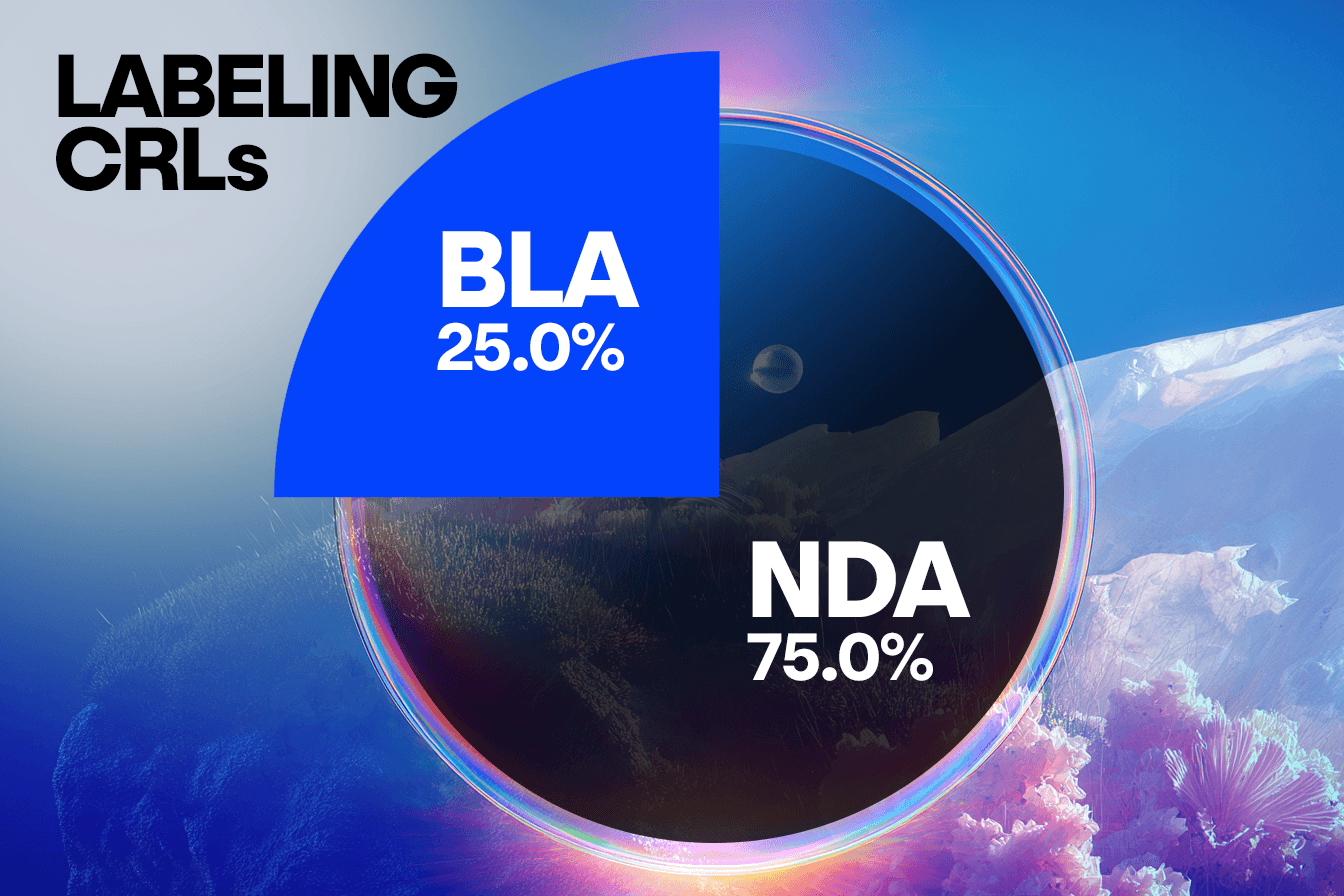
However, a distinct divergence appears in the Other/Regulatory category, where the NDA submissions show a disproportionately higher frequency of concerns compared to the BLAs. This suggests that NDAs may be more prone to missing submission components, having incomplete studies, or facing legal hurdles.
Further investigation is necessary to confirm this hypothesis, as the underlying cause may be related to differences in submission requirements or the number of interactions with the FDA during the review period.
These findings highlight the value of aggregating data to identify unexpected trends that might otherwise go unnoticed.
Additionally, this analysis could suggest how submission processes and requirements may be adjusted to reduce these discrepancies or how companies can be proactive in their conversations with the FDA to ensure they are meeting these requirements.
Navigating Multiple CRLs: Challenges and Trends Across Therapeutic Areas
A strong majority of applications, approximately 75%, achieved approval after resolving the deficiencies identified in just one CRL. However, close to a quarter received more than one, usually two or three, and in a small number of cases, as many as four, five, or even six CRLs before approvals.
Breaking down these multiple CRL cycles across the five most-cited TAs highlights important trends. Oncology stands out prominently, facing the most significant challenges: eight oncology drugs required two CRLs, two required three CRLs, and there were instances where drugs required four, five, or even six CRLs. This indicates that while a considerable number achieve a relatively straightforward path to approval, a larger proportion face repeated regulatory interactions, emphasizing the complexity inherent in oncology drug development.
Conversely, other TAs, such as neurology and pain, also encountered multiple CRL cycles, though less frequently and typically capped at two or three cycles. Cardiovascular and dermatology had proportionally fewer products with multiple CRL cycles.
Additionally, certain issues—particularly clinical, CMC, and labeling/risk management—often reappear in subsequent cycles for applications receiving more than one CRL. This recurrence suggests either that original deficiencies were not fully addressed in initial resubmissions or that inherent complexities related to the drug or therapeutic indication result in persistent regulatory hurdles.
For applications with multiple CRLs, the nature of deficiencies frequently evolves from one cycle to the next. Some drugs initially encounter clinical issues, only to later face CMC or labeling challenges. Others experience persistent difficulties within a single category, indicating ongoing complexities in fully addressing FDA concerns.
The occurrence of multiple CRLs and patterns within them emphasizes the importance of sponsors adopting adaptive, proactive, and cross-functional strategies to respond comprehensively to regulatory feedback.
What Do We Know Now?
The recent release of CRLs offers a rare window into the FDA’s decision-making, giving companies direct access to the agency’s reasoning behind past rejections. This new transparency not only confirms known priorities—like the importance of strong manufacturing practices—but also reveals patterns that can help teams spot and address issues earlier. From clinical gaps to trial design flaws, sponsors now have clearer insight into where others have stumbled—and how to better prepare for a successful submission.
Ultimately, persistence and a collaborative approach are key—even drugs facing multiple rounds of FDA feedback can, and do, succeed in reaching the market.
While the FDA’s CRLs may not be new, the way we can analyze and apply them is. Much of this information has always been available in fragments, buried in sponsor press releases, hinted at in advisory committee transcripts, or redacted in public-facing summaries. What’s changed is our ability to synthesize, scale, and strategically apply these learnings in a way that delivers real value to our clients.
We’ve only scratched the surface of what this data can tell us; there is so much more we can learn, and combining this data with the already-available information will allow us to generate even more insights around timing, approval pathways, product type, and more. The volume and complexity of regulatory information in the life sciences industry continues to grow, creating both a challenge and a strategic opportunity. Success increasingly depends on a team's ability to translate dense, technical language into actionable insight and align early with evolving regulatory expectations.
This is where organizations must shift from reactive interpretation to proactive, insight-driven planning that requires more than just awareness. It calls for deep domain knowledge, scalable tools, and a way to integrate medical, regulatory, and data expertise from the outset.
Recent advances in AI have made it possible to process hundreds of regulatory documents at scale, turning unstructured data into foresight. When paired with subject matter expertise, these tools can help uncover patterns in agency feedback, highlight precedents, and identify early-stage risks before they threaten approvals.
Human interpretation remains critical. AI tools are only as good as the regulatory intelligence behind them, and the ability to connect insights to submission strategies still relies on seasoned judgment. That’s why hybrid models, where automation supports but doesn’t replace expert review, are becoming a preferred approach, particularly in highly regulated environments.

A further advantage comes from institutional memory. Access to internal databases of agency communications, warning letters, and prior pathways can help teams avoid reinventing the wheel. Understanding how similar situations have been interpreted in the past provides valuable context for strategic planning and agency engagement.
And, integration matters. When medical, regulatory, and technical functions work together, it becomes easier to turn insights into cross-functional action. This collaboration can lead to faster turnaround, smarter decision-making, and more resilient regulatory strategies.
Ultimately, in today’s environment, knowing what regulators are saying is no longer enough. The differentiator is knowing what to do next—and that’s where Klick can help.
Klick Health is the world’s largest independent commercialization partner for life sciences and a leading full-service pharma marketing partner, serving as agency of record for leading pharma, biotech, and healthcare brands. Klick’s specialized offerings are rooted in deep medical and scientific understanding, including market insights, award-winning creative, and proprietary AI and data models to craft impactful brand narratives and seamless customer journeys. Backed by nearly 250 medical experts and advanced healthcare analytics, Klick delivers integrated marketing strategy and communications, from new product launch strategy to MLR review with real-world evidence, helping brands thrive in today’s complex healthcare landscape. Learn more at Klick.com.
Authors

Cassandra Carroll, PhD
Director, Science + Regulatory

Kaitlyn Delano, PhD
Director, Science + Regulatory

Kinza Maxood, MD
Director, Science + Regulatory

Vrudhi Shah, MSc
Director, Science + Regulatory

Margot Arntfield, PhD
SVP, Science + Regulatory

JJ Mifsud
Vice President, Applied Artificial Intelligence
Related Content



Ready to Drive Life Sciences Forward?
Experience the transformative power of Klick Health, where deep industry expertise meets cutting-edge AI-driven wisdom.
As your trusted partners in life sciences commercialization, we combine a storied history in healthcare with the latest technologies to elevate every facet of your omnichannel strategy. From crafting engaging narratives to enabling data-driven decision-making, our integrated capabilities ensure you lead the way in transforming patient outcomes through digital health innovation.
Let’s create something transformative together.
